Retrocopies (or processed pseudogenes) are gene copies generated by reverse transcription and genomic reintegration of processed messenger RNAs (mRNAs). This process is mediated by long interspersed nuclear elements (LINEs) acting in trans on mRNA. Since retrocopies originate from processed mRNAs, they may inherit complete/partial ORFs of their parental genes (i.e., the genes providing the mRNA from which retrocopies originate), but they usually lack other gene features such as introns and regulatory elements. Fixed retrocopies are those derived from ancestral retrotransposition events that became fixed in a species. Usually, fixed retrocopies may accumulate mutations, differentiating their genomic sequences from their parent genes, and recruit regulatory elements such as promoters and enhancers to become functional.
Until recently, retrocopies have been referred to as processed pseudogenes, but an increasing body of evidence suggests that a large fraction of retrocopies are functional (Kaessmann et al. 2009; Navarro and Galante 2015; Bim et al. 2019). Now, it is clear that retrocopies are a major source of genetic novelties by creating novel genes (Carelli et al. 2016), regulatory regions (Parker et al. 2009) and other non-coding genes, including miRNAs (Mercuri et al. 2023; Devor 2006). Once retrocopies have the ability to recruit regulatory elements over time and gain expression, studies have demonstrated that retrocopies may have important functional roles(Karreth et al. 2015; Poliseno et al. 2010). For example, retrocopies that have high sequence similarity with their parental genes may code for similar proteins, but functioning in different contexts (e.g., tissues). They may also participate in regulatory processes, acting for example as miRNAs sponges.
RCPedia provides detailed information about more than 219.948 fixed retrocopies in 44 eukaryotic species. RCPedia can be used to extract information about retrocopies coordinates, genomic context (e.g., intragenic, intergenic), nucleotide sequence, conservation and parental genes. RCPedia also provides access to the expression profiles of retrocopies in 154.687 samples from 73 tissues of 15 species, which were obtained from large, public RNA sequencing repositories such as Genotype-Tissue Expression (GTEx; https://gtexportal.org), ARCHS4 (https://maayanlab.cloud/archs4) and Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra).
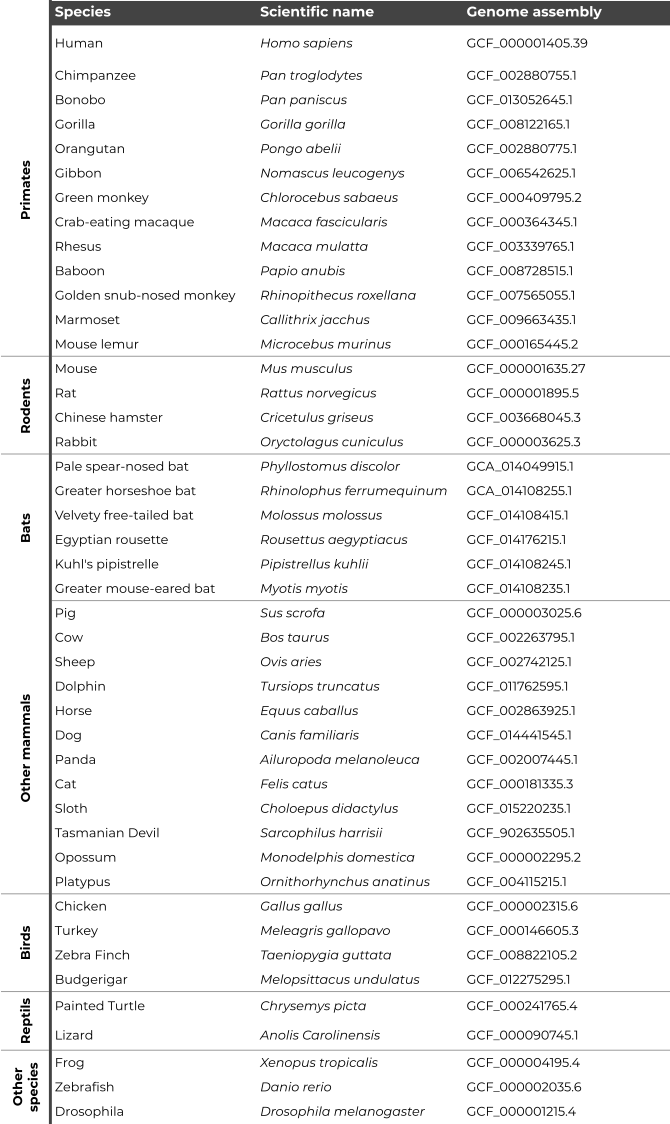
RCPedia includes information about 44 species, encompassing 13 primates, 4 rodents, 6 bats, 12 other mammals, 4 birds, 2 reptiles, and 3 other species. See table below:
Briefly, the identification of fixed retrocopies was performed by first aligning the messenger RNAs (mRNAs) annotated by RefSeq against the reference genome using the LAST aligner(Kiełbasa et al. 2011). Next, we applied a series of scripts in bash, python and perl, in addition to bioinformatics tools such as bedtools to filter the most reliable alignments. First, to remove chromosomal duplications, we selected only the alignments with a match greater than 120 and a distance greater than 200,000 base pairs from the gene of origin of that mRNA. Second, we checked whether the alignment contained the last, penultimate or antepenultimate exon-exon junction of the parental gene, as reverse transcriptase processes mRNAs from the poly-A tail. Third, we verified that these exon-exon junctions were at least 10 base pairs from the beginning or end of our alignment. Fourth, we removed alignments composed mostly of repetitive elements (greater than or equal to 40%) from RepeatMasker (RepeatMasker Home Page ) repetitive region annotation data. Since LAST creates several short and partial alignments, in the next step we had to assemble alignments based on the distance in the genome and RNA, joining alignments from the same mRNA that were up to 6,000 base pairs apart in the genome, allowing for the presence of repetitive elements between the alignments. Candidates that overlap more than 3 exons of annotated coding genes were removed to eliminate false positives (large gene families misidentified as retrocopies). When multiple candidate retrocopies overlapped in the same region and originated from the same parental gene, but from different mRNAs, the candidate with the largest length or the highest score (match/(match+mismatch)) was selected. In case of a tie, we kept all candidates. If the region presented candidates from different genes, a manual analysis of the cases was carried out and only those in which there was clearly a retrocopy insertion within an older retrocopy were kept.
To determine orthologous retrocopies, we first retrieved the genomic sequence surrounding each retrocopy (3000 bp for each side) and performed region alignment against all retrocopy regions (3000 bp for each side) of the other species in a pairwise fashion using the Lastz aligner(Improved pairwise alignment of genomi...). Pairs are filtered based on alignment coverage above 60% and identity above 70% between genetic regions when the comparison is between primates and coverage above 50% and identity above 60% when one or both species are not primates, since we expect a reduced level of conservation. We also verified that at least 60% of the retrocopy was aligned in the “target” region of the other species when the comparison is between primates and at least 50% when one or both species are not primates. In the case of more than one orthology possibility, we select the alignment with the greatest coverage and identity for each backcopy. For reptiles, amphibians, fish and invertebrates (five species) it was not possible to find orthologs due to the low identity and coverage of the alignments.
For human and mouse species, retrocopy read counts were directly obtained from the ARCHS4 repository (https://maayanlab.cloud/archs4) and expression levels in Transcripts Per Million (TPM) were calculated using local R scripts. For human tissues, retrocopy read counts and TPM levels were also directly obtained from the Genotype-Tissue Expression (GTEx; https://gtexportal.org) portal. For the remaining species, SRA samples were selected to represent 6 tissues: Brain, Heart, Kidney, Liver, Ovary and Testis. For 13 species, samples were selected based on (Fukushima and Pollock 2020), while for the remaining a manual selection was made. Gene expression quantification was performed utilizing Kallisto (Bray et al. 2016) with custom build indexes of annotated retrocopies by RCPedia 2.0 and annotated genes by RefSeq(O'Leary et al. 2016).
The home page of RCPedia can be used to search retrocopies by species based on retrocopy name, parental gene name, parental refseq id and ensembl id. Advanced search allows users to search across many species based on list of names, genomic position (inter or intragenic), genomic region, retrocopy size, parental-rtc identity and number of retrocopies per parental gene. Users can also use the Browser web page to access all retrocopies of any desired species.
Conceição HB, Mercuri RLV, de Castro MPM, Ohara DT, Guardia GDA, Galante PAF. RCPedia: A global resource for studying and exploring retrocopies in diverse species. Bioinformatics, Volume 40, Issue 9, September 2024, btae530.
DOI: 10.1093/bioinformatics/btae530